Computational Physics
Electronic structures of metal hydrides and amorphous materials
Metal hydrides and amorphous materials are not only highly important in terms of industrial applications, but are also fascinating systems from a fundamental materials theory point of view. Exploring their rich electronic structure through computational modeling can offer new atomistic insights which could lead to far-reaching developments in experimental materials physics and engineering.
One of the metal hydrides which we investigate is vanadium hydride. The absorption of hydrogen in the vanadium host leads to intriguing geometrical distortions in the latter which in turn affect the dynamics of hydrogen diffusion. We explore this interplay through both molecular dynamics simulations and density functional theory calculations.
Regarding amorphous materials, we have been concentrating on iron zirconium for different mixing ratios of Fe and Zr. The atomistic description of amorphous materials in computational modelling represents a formidable challenge and we have tested a relatively new method which relies on stochastic quenching to reveal new insights into this class of materials.
Metal hydrides and amorphous materials [Link to Ralph’s page]
Contact
Electronic structure of metal hydrides and amorphous materials II
Despite the simplest possible electronic structure of hydrogen, tremendous complexity can arise when it participates in the formation of solids. We have predicted crystal structures of metal hydrides under pressure and utilized the stochastic quenching method to understand the role that hydrogen can play in affecting electronic properties of certain amorphous materials.
We have predicted crystal structures of metal hydrides under pressure to study structural transformation and phenomena such as metallization which allows us to model metallic hydrogen and associated high-temperature superconductivity. Furthermore, very fundamental aspects of hydrogen in metals have been investigated by us, shedding light on the interplay between interstitial sites occupation and lattice strain. Most recently, we have also utilized the stochastic quenching method to improve our understanding of certain amorphous materials and the role that hydrogen can play in affecting their electronic properties.
References
- “A new probe of the electronic structure of Amorphous materials”, M. Strømme, R. Ahuja and G. A. Niklasson, Phys. Rev. Lett. 93 206403 (2004).
- “Cubic metallic phase of aluminum hydride showing improved hydrogen desorption”, R. H. Scheicher, D. Y. Kim, S. Lebègue, B. Arnaud, M. Alouani, and R. Ahuja, Applied Physics Letters 92, 201903 (2008).
- “Crystal structure of the pressure-induced metallic phase of SiH4 from ab initio theory”, D. Y. Kim, R. H. Scheicher, S. Lebègue, J. Prasongkit, B. Arnaud, M. Alouani, and R. Ahuja, Proceedings of the National Academy of Sciences of the USA 105, 16454 (2008).
- “Predicted High-Temperature Superconducting State in the Hydrogen-Dense Transition-Metal Hydride YH3 at 40 K and 17.7 GPa”, D. Y. Kim, R. H. Scheicher, and R. Ahuja, Physical Review Letters 103, 077002 (2009).
- “General trend for pressurized superconducting hydrogen-dense materials”, D. Y. Kim, R. H. Scheicher, H.-K. Mao, T. W. Kang, and R. Ahuja, Proceedings of the National Academy of Sciences of the USA 107, 2793 (2010).
- “Predicted Formation of Superconducting Platinum-Hydride Crystals under Pressure in the Presence of Molecular Hydrogen”, D. Y. Kim, R. H. Scheicher, C. J. Pickard, R. J. Needs, and R. Ahuja, Physical Review Letters 107, 117002 (2011).
- “Unveiling the complex electronic structure of amorphous metal oxides”, C. Århammar, A. Pietzsch, N. Bock, E. Holmström, C.M. Araujo, J. Gråsjö, S. Zhao, S. Green, T. Peery, F. Hennies, S. Amerioun, A. Föhlisch, J. Schlappa, T. Schmitt, V. N. Strocov, G. A. Niklasson, D. C. Wallace, J.E. Rubensson, B. Johansson and R. Ahuja, PNAS (the Proceedings of the National Academy of Sciences, USA) 108, 6355 (2011).
- “Atomic Diffusion in Solid Molecular Hydrogen”, A. B. Belonoshko, M. Ramzan, H. K. Mao and R. Ahuja, Nature-Scientific Reports 3, 2340 (2013).
- “Effect of uniaxial strain on the site occupancy of hydrogen in vanadium from density-functional calculations”, R. Johansson, R. Ahuja, O. Eriksson, B. Hjörvarsson, and R. H. Scheicher, Scientific Reports 5, 10301 (2015).
- “Structural characterization of amorphous Fe(1-x)Zrx”, R. Johansson, G. Muscas, S. George, M. Ahlberg, K. Kádas, D. Arvanitis, R. Ahuja, O. Eriksson, R. H. Scheicher and P. Jönsson, in manuscript.
Contact
Functional Magnetic Materials
Magnetic materials are an essential part of our everyday life. Data storage, communication, and especially green energy production and electrical engines rely on magnetic materials based on high performance magnets which often contain critical components. Huge efforts are made to find suitable non-hazardous replacements. A new emerging field for magnetic materials is magnetic refrigeration. This can be an efficient way to cool environments, both homes as well as fridges and freezers in future.
Today computational simulation plays an inherent role in understanding and optimizing materials properties. We study fundamental properties of magnetic materials for applications in permanent magnets (PM), magnetic cooling (magneto-caloric effect (MCE)), and spintronics based on density functional theory in combination with atomistic spin-dynamics, and Monte Carlo solvers. For PM the key task is to identify new uniaxial phases which have the same characteristics as today's PM but contain much less critical material i.e. rare earth. In case of the MCE fundamental understanding is addressed by investigating materials which show first order (often metamagnetic) magnetic phase transition.
Contact
Superconductivity from 1D electrons
The unique theoretical understanding available for correlated 1D electrons can be leveraged to design superconductors where electron pairing from repulsive interactions is understood, and the critical temperature for the onset of superconductivity could be increased based on accurate theory. This theory can not just be applied to propose novel superconducting devices and bulk materials, but also to understand pairing from repulsive electrons in existing unconventionally superconducting (USC) materials that are built from 1D structures (Bechgaard and Fabre salts, Sr-ladder compounds, K2Cr3As3).
We are developing theory leveraging the unique theoretical understanding available for correlated 1D electrons in order to
- design superconductors where electron pairing from repulsive interactions is understood, and the critical temperature for the onset of superconductivity could be increased based on accurate theory
- design 1D nanowires that are effectively superconducting, and would be so at high temperatures
- understand pairing from repulsive electrons in existing unconventionally superconducting (USC) materials that are built from 1D structures (Bechgaard and Fabre salts, Sr-ladder compounds, K2Cr3As3)
- transfer the mechanism to stabilize superconductivity from 1D to 2D materials, such as doped graphene
Method-wise, these aims will be achieved by combining parallel density matrix renormalization group numerics (pDMRG) with analytical approaches. More information about this work can be found at Theory Group Adrian Kantian.
Background
In condensed matter physics superconducting (SC) materials are of great fundamental and technological interest. In a superconductor, electrons show correlated motion, as opposed to their effectively independent behaviour in a conventional metal – we speak of electron pairing. When the phases of macroscopically many of these pairs become coherent at low enough temperature, they exhibit off-diagonal long range order (ODLRO). This allows for lossless transport of current and energy, which crucially is protected by a SC gap.
The unconventionally superconducting (USC) materials are especially important to condensed matter physics then. As in them pairing results from repulsive electron-electron interactions, they can work at much higher temperatures than conventionally SC materials (where phonon-mediated pairing is just a few Kelvin strong). But no matter which USC material is studied – whether the high-Tc materials, or the organic Bechgaard and Fabre salts (the first materials discovered to be USC) – their fundamental electron pairing mechanism is uniformly very difficult to understand. This prevents theory from guiding any deliberate and systematic increase of critical temperatures for superconductivity in USC materials.
In this context, one-dimensional (1D) electrons have two unique advantages over higher-dimensional ones: in 1D, electron interactions are effectively the strongest, crucial to stability against thermal pair-breaking. And the theory of 1D electrons has a major edge, as it can solve and understand models of USC materials which in higher spatial dimensions remain unsolved, or admit solutions only under strong approximations not widely accepted. The drawback that so far has prevented 1D materials to be used as superconductors are the strong quantum and thermal fluctuations in 1D systems that prevent macroscopic phase coherence of pairs, and thus suppress superconductivity.
This project is based on eliminating this drawback of 1D electron systems via several different mechanisms, each corresponding to concrete physical set-ups. The systems thus enlarged are no longer strictly one-dimensional, but remain in some ways dominated by their original 1D character. The novel theory framework this project is developing will make it possible to gain qualitative and quantitative understanding of both existing and new to-be-proposed USC devices and materials. This understanding will be significantly beyond that currently available for e.g. fully 2D or 3D models and materials, and allow to systematically improve key properties like the critical temperature for superconductivity based on well-controlled and accurate theory.
Oxid Heterostructures
The interfaces of oxide heterostructures provide a strong interplay between spin, orbital, charge and lattice degrees of freedom. Their careful tuning may lead to artificially designed materials with novel properties, opening the possibility to have new design concepts of future nanodevices. Specifically, the interfaces of transition metal oxide heterostructures may give rise to two-dimensional electron gas, charge transfer and charge migration along with complex magnetic phenomena, which can be useful for technological applications.
The structural reconstruction at the interfaces and the surfaces of the transition metal oxides can modify physical properties due to the breaking of the time reversal and/or inversion symmetry. Moreover, the two-dimensionality of the surface reduces the electrons' kinetic energies, thereby enhancing the effects of electron correlations, which can affect many important physical properties such as optical, transport, magnetic and superconductivity, to name a few. Therefore, it is widely recognized that the interfaces of oxide heterostructures present very interesting phenomena, which require fundamental understanding from both experimental and theoretical sides. We employ sophisticated ab initio theories to study electron correlation, transport and complex magnetic structures at these interfaces.
Research
We study the interplay of spin, orbital and lattice degrees of freedom at the interfaces of functional oxide heterostructures. Complex magnetic structures, ferroelectric polarization and effects of electron correlation are investigated at the interfaces by ab initio calculations.
One of our latest achievements is to explain the vertical shift in exchange bias measurements at the heterointerfaces (LSMO/YMnO3).
This work is supported by Carl Tryggers Stiftelse.
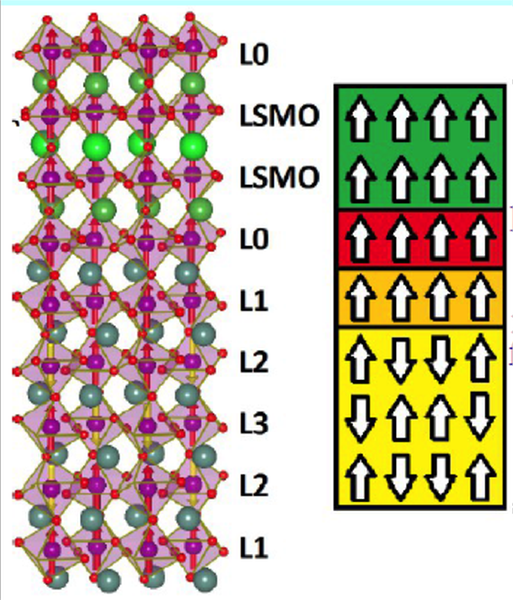
Geometry and magnetic structure of LSMO/YMnO3 interface.
Selected Publications
- Paul et al., J. Appl. Cryst. 47, 1054 (2014)
- Autieri and Sanyal, New J. Phys. 16, 113031 (2014)
- Paul et al., Appl. Phys. Lett. 105, 022409 (2014)
Contact
Dr. Carmine Autieri
Theoretical Spectroscopy
Modern photon and electron beam sources make it possible to probe excitation spectra in many systems, ranging from atoms and molecules to bulk materials and complex interfaces. The interpretation of the experimental data is, however, problematic without an adequate theoretical support. To address this, we develop theories for predicting and analyzing spectra measured in different experiments, as e.g. (angle-resolved) photoemission spectroscopy, magneto-optical spectroscopy, X-ray emission and X-ray absorption, electron energy loss spectra, as well as resonant inelastic X-ray scattering.
The topics of our interest are fundamental theory of spectroscopy, method development, as well as understanding of functional materials, for energy conversion and molecular electronics, or where complex interactions play a role. Examples are Li based batteries and Ru based catalysts for the productions of solar fuels and correlated oxides such as high-temperature superconductors. The theoretical tools we employ involve density functional theory (DFT), linear-response theory, dynamical mean field theory (DMFT), Mahan-Nozieres-De Dominicis theory, and multi-configurational methods used in quantum chemistry.
Name of Barbara’s project
Name of Jan’s project: Electron magnetic circular dichroism
Name of Olle’s project
Name of Peter’s project: Theory of ultrafast magneto-X-ray spectroscopy
Theoretical spectroscopy II
In order to meet the challenges of modern synchrotron radiation facilities, we develop theories for analyzing the experimental results that are produced in these facilities. This involves calculations of x-ray and photoelectron spectra of molecules, interfaces and bulk materials.
The theory is focused not only on functional materials used for instance in energy conversions and molecular electronics, but also on materials in which the fundamental understanding of complex interactions are in focus. Examples of the former are Li based batteries and Ru based catalysts for the productions of solar fuels, and of the latter are correlated oxides such as high-temperature superconductors. The spectra we focus on involve angular resolved photoemission spectroscopy, angle integrated valence band spectra, optical excitations, x-ray emission and x-ray absorption, as well as Resonant Inelastic X-ray Scattering (RIXS). The theoretical tools we employ involve density functional theory, dynamical mean field theory, Mahan-Nozieres-De Dominicis theory, and multi-configurational methods. Examples of this research can be found in Refs. [1]-[6].
References:
- A. Grechnev, I. Di Marco, M. I. Katsnelson, A. I. Lichtenstein, J. Wills and O. Eriksson, “Theory of quasiparticle spectra of Fe, Co and Ni; bulk and surface”, Phys. Rev. B 76, 35107 (2007).
- P. Thunström, I. Di Marco and O. Eriksson, “Electronic Entanglement in Late Transition Metal Oxides”, Phys. Rev Letters 109, 186401 (2012).
- I. Di Marco, P. Thunström, M. I. Katsnelson, J. Sadowski, K. Karlsson, S. Lebegue, J. Kanski, O. Eriksson, “Electron correlations in MnxGa1-xAs as seen by resonant electron spectroscopy and dynamical mean field theory”, Nat. Commun. 4, 2645 (2013).
- B. Brena, C. Puglia, M. de Simone, M. Coreno, K. Tarafder, V. Feyer, R. Banerjee, E. Göthelid, B. Sanyal, P. M. Oppeneer, O. Eriksson, “Valence-Band Electronic Structure of Iron Phthalocyanine: an Experimental and Theoretical Photo Electron Spectroscopy Study”, J. of Chemical Physics 134, 074312 (2011).
- S. Bhowmick, J. Rusz and O. Eriksson, “X-ray absorption spectra: graphene, h-BN and their alloy BC2N”, Phys. Rev. B 87, 155108 (2013).
- R. Sanchez-de-Armas, B. Brena, I. Rivalta and C. Moyses Araujo, “Soft X-ray spectroscopic properties of ruthenium complex catalyst under CO2 electrochemical reduction conditions: a first-principles study”, J. Phys. Chem. C 119, 22899 (2015).
On-surface Magnetochemistry and Spintronics
Spin-bearing metalorganic molecules provide a unique platform for exploiting spin properties, leading to molecular spintronics and on-surface magnetochemistry, an emergent area in which reversible switching of the molecule’s spin is utilized.
To prepare the way for spin-switchable single-molecule electronic devices, we perform fundamental investigations of the complex interplay of chemical and magnetic interactions in metalorganic systems, using density-functional-theory-based methodologies.
Organic molecules are cheap, multifunctional materials that are promising for many technological applications. In the present focus for molecular spintronics applications stand, in particular, magnetic planar organic molecules adsorbed on substrates, such as metal-porphyrins and phthalocyanines and spin-crossover complexes. To investigate how efficient spin manipulation of spin-bearing molecules can be achieved, we use ab initio calculations that can provide valuable, novel insight into the atomic origins of molecular spin polarization and reveal promising routes to achieve active spin control in future molecular devices.
Name of Peter’s project [Link to Peter’s page]
Name of Biplab’s project
Magnetism
We investigate the magnetic properties of transition and rare-earth metals in bulk, multilayer, surface and cluster geometries. We also study the magnetism of novel superconductors, complex oxides and dilute magnetic semiconductors.
Our interest in magnetism is long-standing and extensive. We investigate the magnetic properties of transition and rare-earth metals in bulk, multilayer, surface and cluster geometries. We also study the magnetism of novel superconductors, complex oxides and dilute magnetic semiconductors.
Dynamics
We have recently developed an interest in the dynamical properties of magnetic materials, which is essential for the description of processes such as ultrafast demagnetization.
As part of our work, we have also developed the UppASD code, which we have used, for example, to investigate the properties of ferromagnets in the monolayer limit.
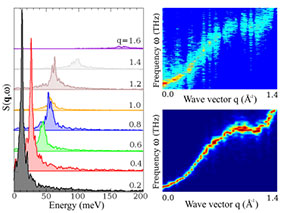
We also employ analytical methods to investigate model Hamiltonians that shed light, for example, on spin-polarized tunneling spectroscopy experiments.
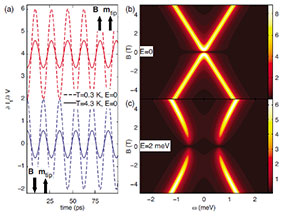
Correlated Electronic Structures
Actinides and transition metal oxides are examples of highly correlated electronic structures. We study such systems using dynamical mean field theory (DMFT), in combination with a full-potential electronic structure method based on linear muffin-tin orbitals (the RSPt code).
The multipole expansion of the Hubbard-U interaction term is explicitly considered in our work based on linear augmented plane-wave method (the Elk code).

Further Reading
- Battiato et al., Superdiffusive Spin Transport as a Mechanism of Ultrafast Demagnetization Phys. Rev. Lett. 105, 027203 (2010).
- Bergman et al., Magnon softening in a ferromagnetic monolayer: A first-principles spin dynamics study Phys. Rev. B 81, 144416 (2010).
- Fransson et al., Theory of spin-polarized scanning tunneling microscopy applied to local spins Phys. Rev. B 81, 115454 (2010).
- Cricchio et al., Itinerant Magnetic Multipole Moments of Rank Five as the Hidden Order in URu2Si2 Phys. Rev. Lett. 103, 107202 (2009).
High moment materials for writer heads
High magnetic moment materials are essential for writer heads of hard disks. This project, in collaboration with Seagate Technology, Ireland and University of Duisburg, Germany, funded by EU deals with the development of high moment materials. The idea is to combine rare earth and transition metal elements to realize large magnetic moments stabilized at high temperatures in a single material.
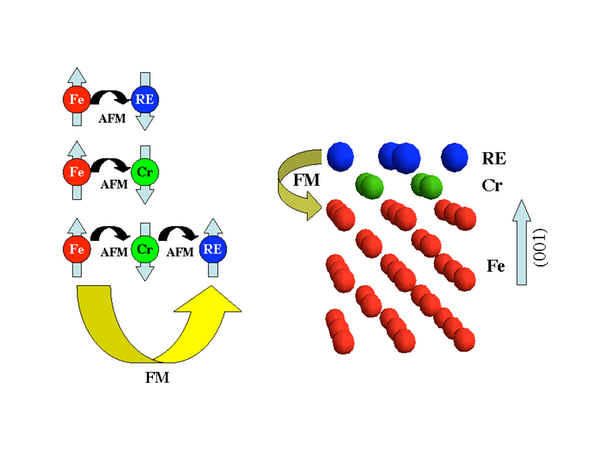
Further reading
- Sanyal et al., Forcing Ferromagnetic Coupling Between Rare-Earth-Metal and 3d Ferromagnetic Films Phys. Rev. Lett. 104, 156402 (2010).
Model Magnetism
Research
The mechanisms of magnetism and magnetic phenomena under non-equilibrium and temporally varying conditions can be efficiently studied using model Hamiltonians. Our primary focus lie on the theoretical description of magnetic effects, especially in nanoscale systems but also in bulk materials. Here, we study the time-dependent interactions between local spin moments and electronic current, and microscopic origin of magnetic interactions, such as spin–electron, spin–spin, spin–lattice, and spin–light interactions.
Special attention is paid to magnetic phenomena that are currently being investigated by local probing techniques (scanning tunneling and atomic force microscopy/spectroscopy). Here we address issues related to the general theoretical description of the measurements but also physical origins of probe induced variations in, e.g., anisotropy fields.
Our work is supported by external grants from the Swedish Research Council and Stiftelsen Olle Engqvist Byggmästare.
Selected publications
- Electrical and Thermal Control of Magnetic Exchange Interactions, J. Fransson, J. Ren, and J.-X. Zhu, Phys. Rev. Lett. 113, 257201 (2014).
- Atomistic Spin Dynamic Method with both Damping and Moment of Inertia Effects Included from First Principles, S. Bhattacharjee, L. Nordström, and J. Fransson, Phys. Rev. Lett. 108, 057204 (2012).
- Dynamical exchange interactions between localized spins out of equilibrium, J. Fransson, Phys. Rev. B, 82, 180411(R) (2010).
- Theory of spin-polarized scanning tunneling microscopy applied to local spins, J. Fransson, O. Eriksson, and A. V. Balatsky, Phys. Rev. B, 81, 115454 (2010).
- Subnanosecond switching of local spin-exchange coupled to ferromagnets, J. Fransson, Phys. Rev. B, 77, 205316 (2008).
Contact
Dr. Simone Borlenghi Garoia
Peter Berggren
Henning Hammar
Juan David Vasquez Jaramillo
Correlated Electronic Structure
For many materials with 3d, 4f or 5f valence electrons the standard computational approach based on density-functional theory (DFT) in local or semi-local approximations fails. In the last decade the state-of-the-art technique to study these systems has become the combination between dynamical mean-field theory (DMFT) and DFT, which is usually addressed as DFT+DMFT. At the Materials Theory Division we have developed our own DFT+DMFT implementation in the full-potential linearized muffin-tin orbital (FP-LMTO) method RSPt. We have used this powerful and flexible code to investigate the electronic structure of several systems, ranging from the late transition metals to their oxides, from dilute magnetic semiconductors to the whole series of the lanthanides.
This project is sponsored by VR, eSSENCE, STANDUPP, and KAW Foundation.
Contact
Weiwei Sun
Johan Schött
Samara Keshavarz
Molecular electronics or Model Magnetism

Molecular electronics is a rapidly developing research field at the interface of physics, chemistry, and engineering, in which electron transport through molecules is investigated. The project involves design and ab initio simulations of molecular structures, metal and semiconductor surfaces and molecular adsorption applied to molecular electronics, biological and nano-sensors and synthesis of novel materials. Our research is performed within the environment of the Uppsala University UniMolecular Electronics Center (U³MEC) which focuses on molecular electronics based on single or small assemblies of molecules.
Nanobiotechnology
Nanobiotechnology is a hot new research field in which nano-sized artificial structures are combined with natural biological systems for a broad range of purposes, among them bio-sensing and drug delivery. We are active in both these research areas.
For sensing applications, our focus is on nanopores as a new approach for sequencing DNA. We use molecular dynamics simulations to explore the movement of DNA in the nanopore channel, density functional theory to determine the electronic structure of the nano-bio device, and quantum transport methods to calculate the electrical conductance which constitutes the output signal to distinguish between the four different bases in DNA.
Our other research focus is the use of 2D nanomaterials as targeted drug delivery agents for the treatment of cancer, antibiotic resistance, neurodegenerative diseases like Alzheimer’s etc. Here, ab initio simulations are used by us to mainly understand the adsorption characteristics of the drug molecules in the context of binding energies, electronic structure and optical properties.
Nanopores for analysis of DNA and proteins [Link to Ralph’s page]
Name of Biplab’s project [Link to Biplab’s page]
On-Surface Magnetochemistry and Spintronics
Spin-bearing metalorganic molecules provide a unique platform for exploiting spin properties, leading to molecular spintronics and on-surface magnetochemistry, an emergent area in which reversible switching of the molecule’s spin is utilized.
To prepare the way for spin-switchable single-molecule electronic devices, we perform fundamental investigations of the complex interplay of chemical and magnetic interactions in metalorganic systems, using density-functional-theory-based methodologies.
Organic molecules are cheap, multifunctional materials that are promising for many technological applications. In the present focus for molecular spintronics applications stand, in particular, magnetic planar organic molecules adsorbed on substrates, such as metal-porphyrins and phthalocyanines and spin-crossover complexes. To investigate how efficient spin manipulation of spin-bearing molecules can be achieved, we use ab initio calculations that can provide valuable, novel insight into the atomic origins of molecular spin polarization and reveal promising routes to achieve active spin control in future molecular devices.
Name of Peter’s project [Link to Peter’s page]
Name of Biplab’s project
On-surface magnetochemistry and spintronics II
Organic molecules are multifunctional materials that are promising for many technological applications. They are cheap materials that can be mass-produced and their functionalities can moreover easily be tailor-made by chemical synthesis. Therefore they are ideally suited for achieving the goal of sustainable development. Spin-bearing metalorganic molecules provide a unique platform for exploiting in addition spin properties, leading to molecular spintronics and on-surface magnetochemistry, an emergent area in which adsorption/desorption of an extraneous molecule is employed to realize reversible switching of the molecule’s spin.
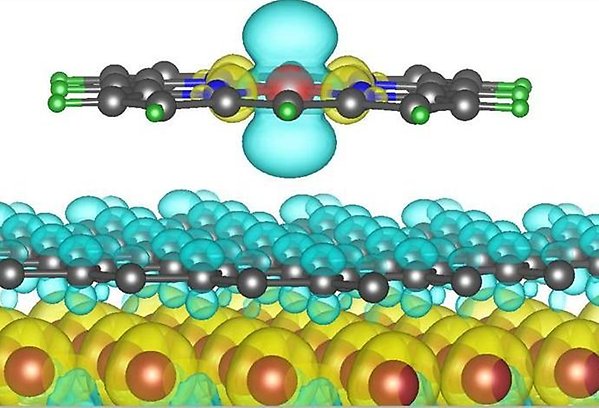
To prepare the way for spin-switchable single-molecule electronic devices, fundamental investigations are required to establish how spin manipulation of spin-bearing molecules can be achieved. To this goal, we use density-functional-theory-based methodologies to study computationally magnetic planar metalorganic molecules adsorbed on substrates, such as porphyrins and phthalocyanines, as well as three-dimensional spin-crossover materials. Our ab initio calculations provide valuable, novel insight into the microscopic origins of molecular spin polarization and reveal promising routes to achieve active control over the spin in future molecular spintronic applications.
Link to On-surface magnetochemistry and spintronics webpage.
Name of Biplab’s project [Link to Biplab’s page]
Ultrafast Magnetic Processes
Ultrafast dynamics in magnetic materials has recently emerged as an intriguing area of modern science. In this field, ultrashort radiation pulses are used to excite a material to a non-equilibrium state that may lead to ultrafast demagnetization, magnetization reversal or even generation of magnetization. The underlying fundamental mechanisms are however poorly understood. To unveil these we develop analytical models and ab initio theory to advance the understanding of ultrafast magnetic processes.
The recent discovery of several unexpected phenomena occurring on sub-picosecond timescales, such as ultrafast demagnetization or transiently induced superconductivity, has emphasized the importance of strongly correlated, out-of-equilibrium processes that involve spin, orbital, and lattice degrees of freedom. Aiming to explain such ultrafast magnetic processes, and prepare the way for their future technological utilization, we develop theory for laser-induced demagnetization, ultrafast spin dynamics and spin reorientation, as well as for non-thermal femtosecond spin currents and transport.
Contact
Contact
- Programme Professor Materials Theory
- Olle Eriksson
- Avdelningsföreståndare
- Biplab Sanyal