Beräkningselektrokemi

Vi arbetar med de grundläggande aspekterna av jonledande lösningar och elektriskt laddade gränssnitt - inom energilagring och omvandling, med beräkningsverktyg.
Enligt den tvådelade boken "Modern Elechrochemistry", skriven av Bockris och Reddy, så finns det två sorters elektrokemi: Den ena är "den fysikaliska kemin av joniskt ledande lösningar" och den andra är "den fysikaliska kemin av elektriskt laddade gränssnitt". Vi arbetar med de grundläggande aspekterna av bägge dessa problem inom elektrokemisk energilagring.
Modellering av elektrokemiska gränssnitt med finit fältmolekylär dynamik
En realistisk representation av ett elektrokemiskt gränssnitt kräver behandling av elektroniska, strukturella och dynamiska egenskaper på lika villkor. Den densitetsfunktionella teoribaserade molekyldynamikmetoden (DFTMD) är kanske det enda tillvägagångssättet som kan ge en konsekvent atomistisk beskrivning. Utmaningen för DFTMD-modellering av materialets gränssnittsdielektrikum är dock den långsamma konvergensen av polarisationen P, där P är en central enhet för att sammanknyta alla dielektriska egenskaper hos ett elektrokemiskt gränssnitt.
Vårt bidrag inom detta område är att utveckla finita fälts-simuleringstekniker för molekyldynamisk modellering av elektrokemiska system [1, 2]. Konstant elektrisk flödestäthet D Hamiltonianen, ursprungligen designad för att behandla spontan polarisering i marktillståndsferroelektriska system, ger en ny statistisk mekanikensemble. Vi visar att fördelen med konstant D-simuleringar inom beräkningselektrokemi är trefaldig: a) Det påskyndar avsevärt simuleringar för både polära vätskor och isolerande oxider; b) Det eliminerar effekten med ändlig storlek för modellering av det elektriska dubbelskiktet på grund av det periodiska gränsvillkoret; c) Den kontrollerar ytladdningstätheten i metalliska elektroder.
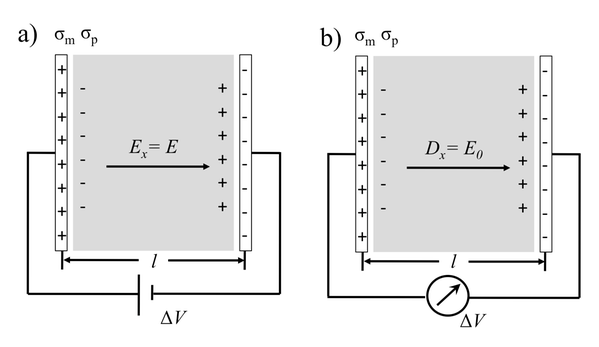
Kredit: Chao Zhang
Jontransport i flytande och polymerelektrolyter
Litiumjonbatterier är elektrokemiska enheter som involverar flera tids- och längdskalor för att uppnå optimal prestanda och säkerhetskrav. När det gäller elektrolyten som fungerar som jonledare är en förståelse på molekylär nivå av motsvarande transportfenomen avgörande för den rationella designen. För närvarande arbetar vi med molekyldynamiksimuleringar av jontransport i olika typer av elektrolyter, från flytande elektrolyter till polymerelektrolyter (med Daniel Brandell), som är relevanta för batteriapplikationer. Vårt bidrag på detta område är att belysa referensramsberoendet vid beräkningen av Onsager-koefficienterna och överföringstalen [3]. Detta steg är avgörande för att maximera kraften i MD-simulering och koppla samman experiment och molekylsimuleringar.
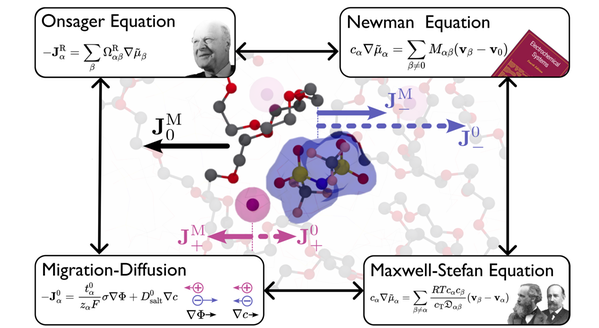
Kredit: Yunqi Shao
Utveckling av atomistisk maskininlärning för elektrokemiska system
Maskininlärning blir allt viktigare i materialmodellering och materialupptäckt. Atomiska neurala nätverk (ANN), som utgör en klass av ML-metoder, har varit mycket framgångsrika i att förutsäga både fysikalisk-kemiska egenskaper och approximera potentiella energiytor.
Nyligen har vi tagit initiativet och utvecklat ett Python-bibliotek med öppen källkod som heter PiNN (https://github.com/Teoroo-CMC/PiNN/), vilket gör det möjligt för forskare att enkelt träna toppmoderna ANN-arkitekturer och implementera nya ML-modeller för att göra kemiska förutsägelser (t.ex. av responsfunktioner). I synnerhet har vi designat och implementerat en tolkbar och högpresterande grafkonvolutionell neural nätverksarkitektur PiNet, och demonstrerar hur den kemiska insikten ett sådant nätverk "lärt sig" kan extraheras [4]. Detta gör att vi kan utföra atomistisk simulering av elektrokemiska system som drivs av ML-modeller, vilket visas av PiNNwall [5].
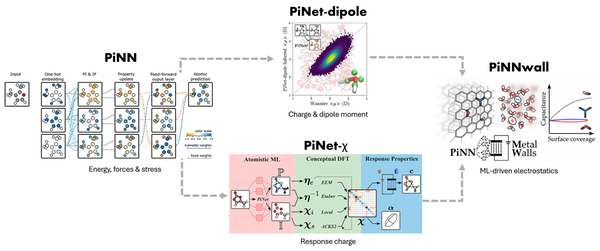
Kredit: Lisanne Knijff
Referenser
- Knijff, J. Mei and C. Zhang, in Encyclopaedia of Solid-Liquid Interfaces., 2024, 567 DOI: 10.1016/B978-0-323-85669-0.00012-X
- Andersson, and C. Zhang, Curr. Opin. Electrochem., 2023, 42: 101407 DOI: doi.org/10.1016/j.coelec.2023.101407
- Shao, H. Gudla, J. Mindemark, D. Brandell and C. Zhang, Acc. Chem. Res., 2024, 57: 1123 DOI: 10.1021/acs.accounts.3c00791
- Shao, L. Knijff, F. M. Dietrich, K. Hermansson and C. Zhang, Batter. Supercaps, 2021, 4: 585 DOI: 10.1002/batt.202000262
- Dufils, L. Knijff, Y. Shao and C. Zhang, J. Chem. Theory Comput., 2023, 19: 5199 DOI: 10.1021/acs.jctc.3c00359
Kontakt
- Om du har några frågor om detta forskningsområde så är du välkommen att kontakta Dr. Chao Zhang.
- Chao Zhang