Nils Welsh
Signalhändelser som deltar i β-cellsdöd, funktion och regenerering.
Både vid typ 1- och typ 2-diabetes finns det en minskning av antalet och funktionen hos β-celler. Orsakerna till ökad död och minskad regenerering och funktion av β-celler är fortfarande okända. Därför är vårt övergripande mål att studera signalhändelser som deltar i β-cellsdöd, funktion och regenerering. Vi fokuserar för närvarande på signalering via tyrosinkinashämmare, ROS-produktion som sker via de NADPH-beroende oxidaserna (NOX) samt adenosin, som allaverkar kontrollera β-cellsdöd och funktion. För dessa ändamål studerar vi mänskliga β-celler (EndoC-betaH1-celler och mänskliga öar) och vi använder tekniker som RNA-seq, CHiP-seq, qPCR, immunoblotanalys, flödescytometri, viral vektormedierad transduktion, siRNA och in vivo modeller.
Betydelsen av tyrosinkinashämmare i ß-cellsapoptos och diabetes
Det har nyligen observerats att patienter som lider av både leukemi och diabetes botades från inte bara leukemi, utan även diabetes, när de behandlades med tyrosinkinashämmaren Imatinib. (Veneri et al., N Engl J Med. 2005 352(10):1049-50). En antidiabetisk verkan av Imatinib vid typ 2-diabetes stöds också av vår observation att Imatinib motverkar insulinresistens inducerad av en diet med hög fetthalt och hyperglykemi hos råttor (Hägerkvist et al., Clinical Science, (Lond). 2008 114(1) :65-71). I en studie från 2009 observerades dessutom att Imatinib inducerade diabetesremission hos db/db-möss, möjligen genom att minska insulinresistens och öka ß-cellmassan (Han et al., Diabetes. 2009 58(2):329 -3). I både djurmodeller och patienter med typ 2-diabetes verkar Imatinib således förbättra den glykemiska kontrollen, möjligen via en insulinsensibiliserande effekt.
Imatinib verkar förebygga och vända inte bara typ 2-diabetes utan även diabetes från djurmodeller med en typ 1-diabetes som liknar en sjukdom. Vi har visat att Imatinib skyddar mot ß-cellsdöd in vitro och förhindrar diabetes hos NOD-möss och streptozotocin-diabetiska möss, båda modellerna för mänsklig ß-cellsdestruktion och typ 1-diabetes (Hagerkvist et al., FASEB J. 2007 feb; 21(2):618-28, Hagerkvist et al., Cell Biol Int. 2006 30(12):1013-7). På senare tid har det observerats av andra att både Imatinib och Sunitinib inte bara förhindrade, utan också reverserade nyuppkomna diabetes hos NOD-möss (Louvet et al., Proc Natl Acad Sci US A. 2008 105(48):18895-900). Det finns alltså principbevis i djurmodeller för en antidiabetisk effekt av Imatinib och liknande tyrosinkinashämmare, och att en begränsad behandlingsperiod inte bara kommer att vända diabetes utan även förmedla långsiktigt skydd mot återutfällning av sjukdomen. Detta har fått oss (Mokhtari och Welsh, Clin Sci (Lond). 2009 118(4):241-7) och andra utredare att föreslå kliniska prövningar där Imatinib ges till patienter med typ 1-diabetes som nyligen debuterat.
Arbetet av andra och oss indikerar att Imatinib motverkar diabetes via olika molekylära mekanismer (Figur 1).
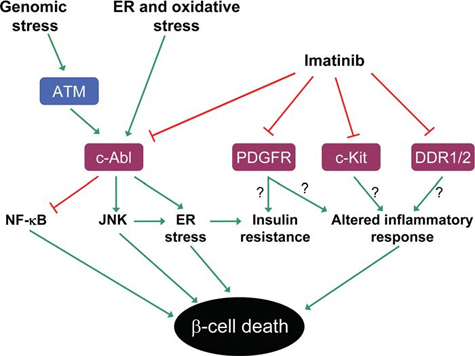
Figur 1. Möjliga mekanismer för de antidiabetiska effekterna av imatinib. Imatinib är känt för att hämma tyrosinkinaserna c-Abl, PDGFR, c-Kit och DDR1/2. Troligtvis förmedlas imatinib-inducerat skydd mot diabetes inte av en enda väg, utan via olika molekylära mekanismer. ß-cellsöverlevnad främjas av hämning av c-Abl, vilket leder till minskad aktivering av det pro-apoptotiska MAPK-kinaset JNK och ökad aktivering av den anti-apoptotiska transkriptionsfaktorn NF-KB. c-Abl-hämning kan också leda till en dämpad ER-stressrespons, via JNK eller andra vägar. Hämning av PDGFR kan bidra till att minska perifer insulinresistens och inflammatoriska processer, och därigenom främja ß-cellöverlevnad. Dessutom kan hämning av c-Kit och DDR1/2 också bidra till de antidiabetiska effekterna av imatinib, möjligen genom att blockera inflammatoriska svar.
Betydelsen av ROS-producerande NADPH-beroende oxidaser (NOX) i β-celldysfunktion
Förlust av pankreas ö-funktion är ett centralt kännetecken i patogenesen av T2DM. Dessutom kan det vara så att även β-cellsförlust inträffar vid T2DM, och att detta börjar, efter en initial fas av hyperinsulinemi, relativt sent i sjukdomsförloppet. Mekanismerna som resulterar i betacellsvikt i T2DM är inte klara, men ackumulerande bevis pekar på en central roll för oxidativ stress som ett resultat av överproduktion av reaktiva syrearter (ROS) (Figur 1).
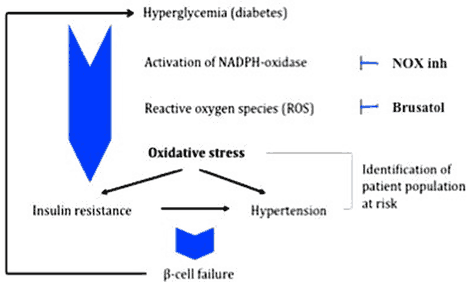
Den överdrivna produktionen och ackumuleringen av ROS beror, åtminstone delvis, på hyperaktivitet av NADPH-oxidaserna (NOX). NOX-familjen består av sju isoformer (NOX1-5 och DUOX1-2), som utför normala cellulära funktioner vid basala förhållanden, men när de ihållande aktiveras producerar skadliga nivåer av ROS. Hyperaktivitet hos några av isoformerna har visat sig vara en viktig drivkraft vid ett antal sjukdomar inklusive diabetes och diabeteskomplikationer [11]. Det aktuella projektet kommer att utforska sätt att skydda mot oxidativ stress och försämring av β-celler genom att hämma NOX, och att definiera T2DM-patientgrupper som särskilt skulle dra nytta av behandling med sådana hämmare. Nya NOX-hämmare är tillgängliga för oss via Glucox Biotech, ett företag som har grundläggande patent, beviljade i USA och i Europa och under behandling i Japan, vilka täcker rätten att utveckla läkemedel mot diabetes som syftar till att hämma NOX. Glucox Biotech äger också internationella (PCT) substanspatentansökningar på sin första och andra generation av substanser.
Publikationer
Ingår i International Journal of Molecular Sciences, 2024
- DOI för GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine
- Ladda ner fulltext (pdf) av GDF15 Protects Insulin-Producing Beta Cells against Pro-Inflammatory Cytokines and Metabolic Stress via Increased Deamination of Intracellular Adenosine

CLEC11A improves insulin secretion and promotes cell proliferation in human beta-cells
Ingår i Journal of Molecular Endocrinology, 2023
- DOI för CLEC11A improves insulin secretion and promotes cell proliferation in human beta-cells
- Ladda ner fulltext (pdf) av CLEC11A improves insulin secretion and promotes cell proliferation in human beta-cells
Ingår i Frontiers in Endocrinology, 2023
- DOI för Metabolic stress-induced human beta-cell death is mediated by increased intracellular levels of adenosine
- Ladda ner fulltext (pdf) av Metabolic stress-induced human beta-cell death is mediated by increased intracellular levels of adenosine
Ingår i Free radical research, s. 460-469, 2023
- DOI för The selective NOX4 inhibitor GLX7013159 decreases blood glucose concentrations and human beta-cell apoptotic rates in diabetic NMRI nu/nu mice transplanted with human islets
- Ladda ner fulltext (pdf) av The selective NOX4 inhibitor GLX7013159 decreases blood glucose concentrations and human beta-cell apoptotic rates in diabetic NMRI nu/nu mice transplanted with human islets
Are off-target effects of imatinib the key to improving beta-cell function in diabetes?
Ingår i Upsala Journal of Medical Sciences, 2022
- DOI för Are off-target effects of imatinib the key to improving beta-cell function in diabetes?
- Ladda ner fulltext (pdf) av Are off-target effects of imatinib the key to improving beta-cell function in diabetes?
Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells
Ingår i Biomedicines, 2021
- DOI för Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells
- Ladda ner fulltext (pdf) av Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells
Ingår i Diabetologia, s. 2292-2305, 2021
- DOI för ZBED6 counteracts high-fat diet-induced glucose intolerance by maintaining beta cell area and reducing excess mitochondrial activation
- Ladda ner fulltext (pdf) av ZBED6 counteracts high-fat diet-induced glucose intolerance by maintaining beta cell area and reducing excess mitochondrial activation
Ingår i Clinical Science, s. 2243-2263, 2021
The importance of the ZBED6-IGF2 axis for metabolic regulation in mouse myoblast cells
Ingår i The FASEB Journal, s. 10250-10266, 2020
- DOI för The importance of the ZBED6-IGF2 axis for metabolic regulation in mouse myoblast cells
- Ladda ner fulltext (pdf) av The importance of the ZBED6-IGF2 axis for metabolic regulation in mouse myoblast cells
Ingår i The FASEB Journal, s. 88-100, 2019
Ingår i The FASEB Journal, s. 3510-3522, 2019
Ingår i Proceedings of the National Academy of Sciences of the United States of America, 2018
The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro
Ingår i PLOS ONE, 2018
- DOI för The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro
- Ladda ner fulltext (pdf) av The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro
Ingår i Islets, s. 43-48, 2017
Department of Medical Cell Biology: Annual Report 2016
Uppsala University, 2017
Ingår i Upsala Journal of Medical Sciences, s. 149-159, 2017
- DOI för Addition of exogenous sodium palmitate increases the IAPP/insulin mRNA ratio via GPR40 in human EndoC-beta H1 cells
- Ladda ner fulltext (pdf) av Addition of exogenous sodium palmitate increases the IAPP/insulin mRNA ratio via GPR40 in human EndoC-beta H1 cells
Ingår i Diabetologia, 2016
Imatinib prevents beta cell death in vitro but does not improve islet transplantation outcome
Ingår i Upsala Journal of Medical Sciences, s. 140-145, 2016
- DOI för Imatinib prevents beta cell death in vitro but does not improve islet transplantation outcome
- Ladda ner fulltext (pdf) av Imatinib prevents beta cell death in vitro but does not improve islet transplantation outcome
Department of Medical Cell Biology: Annual Report 2015
Uppsala University, 2016
Ingår i Heliyon, 2016
- DOI för PTB and TIAR binding to insulin mRNA 3'- and 5'UTRs; implications for insulin biosynthesis and messenger stability.
- Ladda ner fulltext (pdf) av PTB and TIAR binding to insulin mRNA 3'- and 5'UTRs; implications for insulin biosynthesis and messenger stability.
Ingår i European Journal of Pharmacology, s. 69-80, 2016
Ingår i Scientific Reports, 2016
- DOI för Knock-down of ZBED6 in insulin-producing cells promotes N-cadherin junctions between beta-cells and neural crest stem cells in vitro
- Ladda ner fulltext (pdf) av Knock-down of ZBED6 in insulin-producing cells promotes N-cadherin junctions between beta-cells and neural crest stem cells in vitro
Ingår i Upsala Journal of Medical Sciences, s. 40-46, 2015
Ingår i Clinical Science, s. 17-28, 2015
Ingår i Free radical research, s. 1308-1318, 2015
- DOI för The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice
- Ladda ner fulltext (pdf) av The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice
Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro
Ingår i Biochemical and Biophysical Research Communications - BBRC, s. 868-872, 2015
- DOI för Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro
- Ladda ner fulltext (pdf) av Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro
Department of Medical Cell Biology: Annual Report 2014
Uppsala universitet, 2015
Role of the AMP kinase in cytokine-induced human EndoC-beta H1 cell death
Ingår i Molecular and Cellular Endocrinology, s. 53-63, 2015
- DOI för Role of the AMP kinase in cytokine-induced human EndoC-beta H1 cell death
- Ladda ner fulltext (pdf) av Role of the AMP kinase in cytokine-induced human EndoC-beta H1 cell death
Activated pancreatic stellate cells can impair pancreatic islet function in mice
Ingår i Upsala Journal of Medical Sciences, s. 169-180, 2015
Ingår i Pancreas, s. 624-629, 2014
Ingår i Upsala Journal of Medical Sciences, s. 306-315, 2014
- DOI för Bcl-2 maintains the mitochondrial membrane potential, but fails to affect production of reactive oxygen species and endoplasmic reticulum stress, in sodium palmitate-induced beta-cell death
- Ladda ner fulltext (pdf) av Bcl-2 maintains the mitochondrial membrane potential, but fails to affect production of reactive oxygen species and endoplasmic reticulum stress, in sodium palmitate-induced beta-cell death
Ingår i Genomics, s. 264-275, 2014
Ingår i Transplantation, 2013
Ingår i Diabetologia, s. 1327-1338, 2013
- DOI för Imatinib mesilate-induced phosphatidylinositol 3-kinase signalling and improved survival in insulin-producing cells: role of Src homology 2-containing inositol 5'-phosphatase interaction with c-Abl
- Ladda ner fulltext (pdf) av Imatinib mesilate-induced phosphatidylinositol 3-kinase signalling and improved survival in insulin-producing cells: role of Src homology 2-containing inositol 5'-phosphatase interaction with c-Abl
Ingår i PLOS ONE, 2013
- DOI för Co-Culture of Neural Crest Stem Cells (NCSC) and Insulin Producing Beta-TC6 Cells Results in Cadherin Junctions and Protection against Cytokine-Induced Beta-Cell Death
- Ladda ner fulltext (pdf) av Co-Culture of Neural Crest Stem Cells (NCSC) and Insulin Producing Beta-TC6 Cells Results in Cadherin Junctions and Protection against Cytokine-Induced Beta-Cell Death
Ingår i Proceedings of the National Academy of Sciences of the United States of America, s. 15997-16002, 2013
Ingår i Cell Death and Differentiation, s. 1836-1846, 2012
Ingår i Pancreas, s. 490-492, 2012
Ingår i Biochemical and Biophysical Research Communications - BBRC, s. 845-850, 2012
- DOI för Cytokine-induced human islet cell death in vitro correlateswith a persistently high phosphorylation of STAT-1, but not with NF-κB activation
- Ladda ner fulltext (pdf) av Cytokine-induced human islet cell death in vitro correlateswith a persistently high phosphorylation of STAT-1, but not with NF-κB activation
Ingår i Expert Opinion on Investigational Drugs, s. 1743-1750, 2012
- DOI för Does the small tyrosine kinase inhibitor imatinib mesylate counteract diabetes by affecting pancreatic islet amyloidosis and fibrosis?
- Ladda ner fulltext (pdf) av Does the small tyrosine kinase inhibitor imatinib mesylate counteract diabetes by affecting pancreatic islet amyloidosis and fibrosis?
The human insulin mRNA is partly translated via a cap- and eIF4A-independent mechanism
Ingår i Biochemical and Biophysical Research Communications - BBRC, s. 693-698, 2011
Ingår i PLOS ONE, 2011
Ingår i PLOS ONE, 2010
Ingår i Textbook of Diabetes, s. 1064-1069, Wiley-Blackwell, 2010
Potential utility of small tyrosine kinase inhibitors in the treatment of diabetes
Ingår i Clinical Science, s. 241-247, 2010
Ingår i Biochemical and Biophysical Research Communications - BBRC, s. 553-557, 2009
The importance of RNA binding proteins in preproinsulin mRNA stability
Ingår i Molecular and Cellular Endocrinology, s. 28-33, 2009
Ingår i American Journal of Physiology. Endocrinology and Metabolism, 2009
Detailed transcriptome atlas of the pancreatic beta cell
Ingår i BMC Medical Genomics, s. 3, 2009
Ingår i Biochemical Pharmacology, s. 1748-1756, 2008